We began
with a scientific understanding that Pulmonary Fibrosis is a disease of aging. At
one point, he said that if we all lived to 150 years old, we would all have
pulmonary fibrosis. Why? Here we go:
He
primarily talked about Idiopathic Pulmonary Fibrosis. That means that the
origin of the disease is unknown but the results are just like all of us with
an interstitial lung disease: our lungs become fibrotic. So, in effect, the
process of IPF is very similar to any other scaring in an ILD.
We
reviewed the basis structure of the lungs beginning with the trachea then the
branches in the lungs grow smaller with 25-35 connectors to end at the alveoli,
which looks like sponges. This is where the gasses are exchanged. They are
three cells thick – blood cell, epithelial cell, fibroblasts.
With
fibrosis, these areas fill with scar tissue, grow thicker and the transference
of gasses is not properly achieved.
Dr.
Wolters explained that research into the disease of Dyskeratosis Congenita gave
the first clue to understanding fibrosis. It is a disease of premature aging.
The patient’s skin becomes hyperpigmentation and they also have naidstrophy,
oral leukoplakia, liver cirrhosis and 20-30% get lung fibrosis. They discovered
a genetic mutation in this group of people. It had to do with telomeres and
telomerase. Telomeres are the aging agent. They began to see that the telomeres
were damaged in patients with fibrosis.
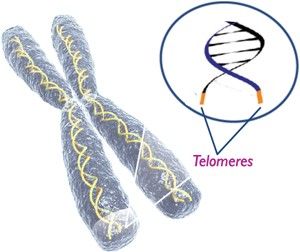
Telomeres
are part of the DNA. A chromosome is just a long string of molecules. It has
ends, which are protected by proteins. They act like a shoelace with the ends
that protect so that the laces don’t become ragged. The same is true of the
proteins that surround the ends of the chromosome. If something happens and the
ends of the chromosome are not protected, they grow shorter. Everyone’s
chromosomes grow shorter getting older thus is the reason we age. It all has to
do with the length of the telomeres. Aging is programmed. In the Dyskeratosis
Congenita, the proteins fall off as the cells are constantly being regenerated,
which is why they age faster than normal.
So,
cells divide, the telomeres get shorter and shorter and when critical, it dies.
This is aging.
So, a
lung doctor studied familial ILD (within a family) for the genetic link. They
related to the telomeres and sequenced the gene.
The
familial IPF studies showed 10% had a mutated gene but all familial IPF
patients had short telomeres. Very, very few people are diagnosed with IPF
under the age of 50. It happens when the telomeres have become shorter and
shorter.
The next
study was with non-familial IPF also had short telomeres. We actually saw
staining on lab slides of lung tissue and were able to see the telomeres. How
is it measured? With a blood test. The shorter the telomeres, the more advanced
the lung disease.
Short
telomeres + time = lung fibrosis
Mom and
Dad both contribute to a child. One may have shorter telomeres. That one is the
weakest link. It is passed on to the next generation. The following generations
will get even shorter telomeres until finally, the two shortest chromosomes
drive the process. Families with mutations can develop different forms
depending on age, health and environments. He also mentioned that they feel
that IPF may in fact be caused by an exposure to a dusty working environment…
Someone
asked Dr. Wolters his goal as a researcher. He said that he is trying to understand
the disease and trying to find a way to slow it down. He mentioned that
cirrhosis of the liver also related to IPF in their research.
Stem
cells are a whole other ballgame. They are in us; we can culture them in a
dish. Many tissues and organs have stem cells. Stem cell failure leads to the
inability of lungs to heal and for fibrosis to occur.
To
engineer new lungs in a rat, researchers observe the epithelial cells in air sac; the air sacs
sit on proteins in a matrix with a few fibroblasts and all rest on blood
vessels. Researchers washed the lungs with a soap to get rid of the cells but
kept the matrix of protein. It looked like a lung but it had no cells. They added
epithelial cells into the air sacs and the lungs worked in a rat. The structure
was the same. It grew and worked…only for 8 hours.
Through
this process of washing lungs, they are taking human lungs, which are injured,
wash and heal them for transplantation into people. “Lung in a Box.” They are
reconditioning what were formerly non-transplantable lungs. The next step will
be fixing the patients own matrix.
Dr.
Wolters listed the following at the end:
Clinical
studies are important especially those using biological samples from patients
Animal
models are vital
IPF is a
disease of the aging
Lungs
likely have cells capable of cell renewal (stem cell)
Using
stem cells to reengineer or treat lung disease is at the embryonic development
phase.
Future: Stem cell magic is a long way off for
lung disease. What is happening right now is that drug companies are developing
drugs to make the telomeres longer or try to stop them from becoming shorter.
If the telomeres stay longer, aging is slowed and diseases are postponed.
I hope I
was able to accurately relate what was presented. Just when I thought I understood what happened to me and why
I got this disease, I realized today there is oh so much more to learn.
No comments:
Post a Comment